
Plik Zespół Jouberta to zaburzenie pochodzenia genetycznego, które charakteryzuje się spadkiem napięcia mięśniowego, problemami z koordynacją, nieprawidłowymi ruchami oczu, zmienionymi wzorcami oddechowymi i niepełnosprawnością intelektualną (Fundacja Joubert Syndrome, 2016).
Wszystkie te zmiany wynikają z autosomalnej transmisji genetycznej, która spowoduje poważne nieprawidłowości w mózgu, redukcję robaka móżdżku, a także nieprawidłowości w strukturze pnia mózgu (National Institute of Neurological Disorders and Stroke, 2016).
Ponadto zespół Jouberta należy do grupy zaburzeń zwanych ciliopatiami, które obejmują dysfunkcję części komórek zwanych rzęskami. Fundacja Zespołu Jouberta, 2016).
Wstępnego opisu tej patologii dokonali Marie Joubert i wsp. W 1968 roku, w którym opisano cztery przypadki. U pacjentów występował częściowy lub całkowity brak robaka móżdżku, epizodyczny zespół ampnea-hypernea u noworodków, nieprawidłowe ruchy oczu, ataksja i upośledzenie umysłowe (Angemi i Zucotti, 2012)..
Ponadto zespół ten był również związany z różnymi zmianami wielonarządowymi, takimi jak zwłóknienie wątroby, polidaktylia, nefronoptyoza lub dystrofia siatkówki (Angemi i Zucotti, 2012).
Jeśli chodzi o leczenie, obecnie nie ma lekarstwa na zespół Jouberta. Interwencje terapeutyczne mają na celu kontrolę i wsparcie objawowe, stymulację fizyczną i intelektualną dzieci oraz terapię zajęciową (National Institute of Neurological Disorders and Stroke, 2016).
Indeks artykułów
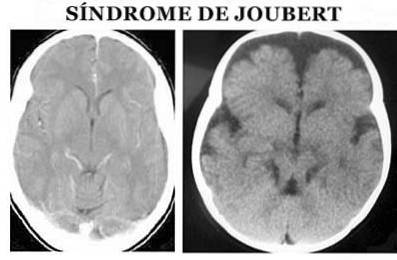
Zespół Jouberta (JS) to rodzaj patologii pochodzenia genetycznego, który charakteryzuje się wrodzonymi wadami rozwojowymi w obszarach pnia mózgu i agenezą (częściową lub całkowitą nieobecność) lub hipoplazją (niepełnym rozwojem) robaka móżdżku, co może powodować (Ophatnet , 2009).
Mówiąc dokładniej, na poziomie anatomicznym charakteryzuje się tzw.znakiem trzonowym śródmózgowia: agenezją lub hipoplazją robaka móżdżku, zwężeniem górnych szypułek móżdżku z pogrubieniem, wydłużeniem i brakiem odkrztuszania oraz głębokim dołem międzykręgowym (angemi i Zuccoti, 2012).
Jest to zaburzenie, które może wpływać na wiele obszarów i narządów ciała, więc objawy przedmiotowe i podmiotowe różnią się znacznie u osób dotkniętych chorobą (U.S. National Library of Medicine, 2011).
Większość osób dotkniętych chorobą cierpi na osłabienie napięcia mięśniowego (hipotonia) i problemy z koordynacją ruchową (ataksja). Inne charakterystyczne cechy to: epizody zmienionego oddychania, oczopląs (mimowolne i arytmiczne ruchy oczu), opóźniony rozwój motoryczny i zmienne trudności intelektualne (US National Library of Medicine, 2011).
Częstość występowania zespołu Jouberta szacuje się na około 1/80 000 do 1/100 000 000 żywych urodzeń. Na całym świecie zarejestrowano ponad 200 przypadków klinicznych (Angemi i Zuccoti, 2012).
Wielu specjalistów uważa te liczby za niedoszacowane, ponieważ zespół Jouberta ma szeroki zakres afektów i jest powszechnie niedodiagnozowany (U.S. National Library of Medicine, 2011).
Wiele klinicznych objawów zespołu Jouberta jest bardziej niż ewidentnych w dzieciństwie, wiele dzieci dotkniętych chorobą ma znaczne opóźnienia ruchowe (National Organization for Rare Disease, 2011).
Najczęstszymi cechami przebiegu klinicznego są: brak kontroli mięśni (ataksja), zmienione wzorce oddechowe (hiperkapnia), bezdech senny, nieprawidłowe ruchy oczu (oczopląs) i niskie napięcie mięśni (National Organization for Rare Disease, 2011).
Z drugiej strony, niektóre ze zmian, które mogą być związane z zespołem Jouberta, obejmują: zmieniony rozwój siatkówki, nieprawidłowości w tęczówce, zez, zmiany w nerkach i / lub wątrobie, wysunięcie błon pokrywających mózg, między innymi ( Krajowa Organizacja Rzadkich Chorób, 2011).
Wszystkie zmiany wynikające z tego zespołu obejmują kilka obszarów: neurologiczne, oczne, nerkowe i mięśniowo-szkieletowe (Bracanti et al., 2010).
Najbardziej charakterystyczne zmiany neurologiczne zespołu Jouberta to Bracanti i wsp., 2010): hipotonia, ataksja, uogólnione opóźnienie rozwoju, zmiany intelektualne, zmiany wzorców oddechowych i nieprawidłowe ruchy oczu.
Na poziomie fizycznym siatkówka jest jednym z narządów dotkniętych zespołem Jouberta. Zmiany w tym narządzie przejawiają się w postaci dystrofii siatkówki, na skutek postępującej degeneracji komórek odpowiedzialnych za fotodbiór.
Klinicznie, zmiany oczne mogą obejmować zarówno wrodzoną ślepotę siatkówki, jak i postępującą degenerację siatkówki.
Z drugiej strony można również zaobserwować obecność colobomy. Ta zmiana oka jest wrodzoną wadą, która wpływa na tęczówkę oka i objawia się jako dziura lub szczelina.
Patologie związane z czynnością nerek dotykają ponad 25% osób dotkniętych zespołem Jouberta.
W wielu przypadkach zaburzenia czynności nerek mogą pozostawać bezobjawowe przez kilka lat lub mogą się objawiać niespecyficznymi objawami, aż przejawiają się ostrą lub przewlekłą niewydolnością nerek..
Od pierwszych opisów tej patologii częstym objawem klinicznym jest polidaktialia (zaburzenie genetyczne, które zwiększa liczbę palców rąk i nóg).
Ponadto często obserwuje się anomalie ustno-twarzowe lub strukturalne na poziomie kręgosłupa.
Badania eksperymentalne sklasyfikowały zespół Jouberta jako zaburzenie autosomalne recesywne (National Organization for Rare Disease, 2011).
Choroba genetyczna autosomalna recesywna oznacza, że dwie kopie nieprawidłowego genu muszą być obecne, aby cecha lub choroba wystąpiła (National Institutes of Health, 2014).
Dlatego recesywna zmiana genetyczna występuje, gdy osoba dziedziczy ten sam nieprawidłowy gen dla tej samej cechy od każdego z rodziców. Jeśli dana osoba otrzyma tylko jedną kopię genu związanego z chorobą, będzie nosicielem, ale nie będzie wykazywać objawów (National Organization for Rare Disease, 2011).
Ponadto co najmniej dziesięć genów zidentyfikowano jako jedną z możliwych przyczyn zespołu Jouberta (National Organization for Rare Disease, 2011).
Mutacja w genie AHI1 jest odpowiedzialna za ten stan patologiczny u około 11% rodzin dotkniętych chorobą. U osób z tą zmianą genetyczną zmiany widzenia są częste z powodu rozwoju dystrofii siatkówki (National Organization for Rare Disease, 2011).
Mutacja genu nphp1 jest przyczyną około 1-2% przypadków zespołu Jouberta. U osób z tą zmianą genetyczną często występują zmiany w nerkach (National Organization for Rare Disease, 2011).
Z drugiej strony mutacja genu CEP290 jest przyczyną 4-10% przypadków zespołu Jouberta (National Organization for Rare Disease, 2011).
Ponadto mutacje w genach TME67, JBTS1, JBTS2, JBTS7, JBTS8 i JBTS9 są również związane z rozwojem zespołu Jouberta (National Organization for Rare Disease, 2011).
Rozpoznanie zespołu Jouberta dokonywane jest na podstawie objawów fizycznych. Konieczne jest zarówno szczegółowe badanie fizykalne, jak i zastosowanie różnych testów diagnostycznych, zwłaszcza obrazów rezonansu magnetycznego (Ophatnet, 2009).
Ponadto molekularne testy genetyczne są również często wykorzystywane do identyfikacji zmian genetycznych, które zostały wykazane w 40% przypadków zespołu Jouberta (National Organization for Rare Disease, 2011).
Z drugiej strony możliwe jest również postawienie prenatalnej diagnozy tej patologii za pomocą USG płodu i analizy molekularnej, szczególnie w rodzinach z genetycznym wywiadem zespołu Jouberta (Ophatnet, 2009).
Gdy najbardziej charakterystyczne cechy zespołu Jouberta występują w połączeniu z jedną lub kilkoma dodatkowymi patologiami fizycznymi, można postawić diagnozę zespołu Jouberta i powiązanych zaburzeń (JSRD) (US National Library of Medicine, 2011).
Dlatego w zależności od rodzaju pokrewnej patologii związanej z obecnością zespołu Jouberta możemy znaleźć podtypy tego. Jednak system klasyfikacji zespołu Jouberta nadal znajduje się w fazie ewolucji ze względu na odkrycie wkładu genetycznego i większą wiedzę na temat korelacji fenotypowych..
Możemy zatem znaleźć (Bracanti i in., 2010):
Leczenie stosowane w zespole Jouberta jest objawowe i podtrzymuje podstawowe patologie. Oprócz interwencji farmakologicznych często stosuje się wczesną stymulację fizyczną i poznawczą (National Institute of Neurological Disorders and Stoke, 2016).
Gdy zmiany oddechowe są znaczące, zwłaszcza w początkowych fazach życia, konieczne jest monitorowanie czynności układu oddechowego (National Institute of Neurological Disorders and Stoke, 2016).
Z drugiej strony identyfikacja i kontrola zwyrodnienia oka, powikłań nerkowych i pozostałych powikłań związanych z zespołem Jouberta powinny być przeprowadzone jak najwcześniej w celu dostosowania środków terapeutycznych (National Institute of Neurological Disorders and Stoke, 2016 ).


Jeszcze bez komentarzy