
Plik Zespół Williamsa jest to zaburzenie rozwojowe pochodzenia genetycznego, które wiąże się z charakterystycznym profilem upośledzeń fizycznych i poznawczych. W szczególności na poziomie klinicznym charakteryzuje się 4 punktami kardynalnymi: 1) nietypowe rysy i cechy twarzy, 2) uogólnione opóźnienie rozwoju psychomotorycznego i specyficznego profilu neurokognitywnego, 3) zmiany sercowo-naczyniowe oraz t) możliwość wystąpienia hiperkalcemii niemowlęcej.
Pomimo faktu, że zespół Williamsa jest uważany za rzadką chorobę, na całym świecie są tysiące osób dotkniętych tą chorobą. Jeśli chodzi o diagnozę, badanie kliniczne zwykle dostarcza niezbędnych ustaleń do jej ustalenia, jednak aby wykluczyć inne patologie i fałszywie dodatnie, zwykle rozpoczyna się badanie genetyczne za pomocą różnych technik..
Z drugiej strony nie ma lekarstwa na zespół Williamsa ani standardowego protokołu leczenia, więc większość interwencji terapeutycznych będzie próbowała regulować powikłania medyczne. Ponadto konieczne będzie uwzględnienie w interwencjach programów wczesnej opieki, zindywidualizowanej edukacji specjalnej i stymulacji neuropsychologicznej..
Indeks artykułów
Zespół Williamsa to zaburzenie rozwojowe, które może znacząco wpływać na różne obszary.
Ogólnie patologia ta charakteryzuje się obecnością nietypowych rysów twarzy lub zmian sercowo-naczyniowych, umiarkowaną niepełnosprawnością intelektualną, problemami w nauce i charakterystycznymi cechami osobowości..
Tak więc pierwszy pacjent z zespołem Williamsa został opisany przez dr Guido Fanconiego w raporcie klinicznym z 1952 r. Jednak to kardiolog Joseph Williams w 1961 r. Precyzyjnie zidentyfikował tę patologię, a także został opisany przez niemieckiego Beurena..
Z tego powodu zespół Williamsa otrzymuje swoją nazwę od obu autorów (zespół Williamsa-Beurena) lub po prostu od pierwszego.
Pomimo tego, że jeszcze kilka lat temu identyfikację patologii prowadzono na podstawie cech fenotypowych, w 1993 roku Edward i wsp. Jako przyczynę etiologiczną stwierdzili nieprawidłowość genetyczną w chromosomie 7q 11.23..
Chociaż zespół Williamsa wiąże się z wieloma różnymi wtórnymi powikłaniami medycznymi, nie ma wysokiej śmiertelności. W wielu przypadkach osoby dotknięte chorobą są w stanie osiągnąć niezależny poziom funkcjonalny.
Zespół Williamsa jest uważany za rzadkie lub rzadkie zaburzenie genetyczne.
Stowarzyszenie Williams Syndrome Association, między innymi, oszacowało, że zespół Williamsa występuje na świecie na około 1 przypadek na 10 000 osób. W szczególności stwierdzono, że w Stanach Zjednoczonych może być dotkniętych około 20 000 lub 30 000.
Jeśli chodzi o rozmieszczenie patologii według płci, nie ma aktualnych danych wskazujących na wyższą częstość występowania w którymkolwiek z nich, ponadto nie zidentyfikowano różnic między regionami geograficznymi lub grupami etnicznymi.
Z drugiej strony wiemy również, że zespół Williamsa jest sporadycznym schorzeniem, chociaż opisano niektóre przypadki transmisji rodzinnej.
Zespół Williamsa, podobnie jak inne patologie pochodzenia genetycznego, ma przebieg kliniczny charakteryzujący się zajęciem wieloukładowym.
Wielu autorów, takich jak González Fernández i Uyaguari Quezada, opisuje kliniczne spektrum zespołu Williamsa podzielone na kilka obszarów: między innymi cechy biomedyczne, cechy psychomotoryczne i poznawcze, cechy psychologiczne i behawioralne..
Fizyczna afektacja obecna w zespole Wiliamsa jest zróżnicowana, wśród najczęstszych objawów klinicznych, jakie możemy zaobserwować:
Opóźniony lub spowolniony rozwój można wykryć już w czasie ciąży. Dzieci dotknięte zespołem Williamsa często rodzą się z niską wagą i wzrostem. Ponadto po osiągnięciu stadium dorosłego całkowita wysokość jest zwykle niższa niż w populacji ogólnej, około 10-15 cm.
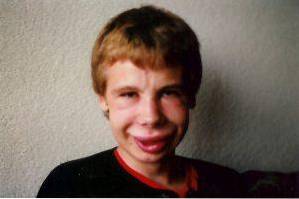
Zmiany twarzy są jednym z najbardziej charakterystycznych objawów klinicznych tego zespołu. U osób dotkniętych chorobą obserwuje się wyraźnie wąskie czoło, zaznaczone fałdy skórne w szparach powiekowych, zez, tęczówkę gwiaździstą, krótki i spłaszczony nos, wydatne kości policzkowe i mniejszą niż zwykle brodę..
W przypadku zmian związanych z rozwojem mięśni i kości można zaobserwować m.in. zmniejszenie napięcia i siły mięśniowej, wiotkość stawów, skoliozę, przykurcze. Wizualnie można zaobserwować postawę charakteryzującą się opadającymi ramionami i częściowo zgiętymi kończynami dolnymi..
Chociaż zwykle nie ma znaczących nieprawidłowości ani wad rozwojowych w małżowinie usznej, we wszystkich przypadkach rozwija się wzrost wrażliwości słuchowej. Osoby dotknięte chorobą mają tendencję do postrzegania lub odczuwania pewnych dźwięków jako irytujących lub bolesnych.
Skóra zwykle jest mało elastyczna, więc można zaobserwować wczesne oznaki starzenia. Ponadto mogą rozwinąć się przepukliny, szczególnie w okolicy pachwinowej i pępkowej..
Różne nieprawidłowości w sercu i naczyniach krwionośnych stanowią najpoważniejsze powikłanie medyczne, ponieważ mogą zagrozić życiu chorego.
Wśród anomalii sercowo-naczyniowych do najczęstszych należą nadkomorowe zwężenie aorty, zwężenie gałęzi płucnej, zwężenie zastawki aortalnej. Wszystkie te zmiany na poziomie klinicznym mogą wpływać na inne obszary naczyniowe, a nawet mózg, ze względu na rozwój nadciśnienia tętniczego.
Nieprawidłowości związane z czynnością nerek i pęcherza są bardzo częste. Ponadto można również wykryć nagromadzenie wapnia (nefrokalcynoza), nagłe parcie na mocz lub moczenie nocne.
Na poziomie poznawczym najważniejszymi cechami są uogólnione opóźnienie w nabywaniu zdolności motorycznych, umiarkowane opóźnienie intelektualne oraz różne zmiany związane z percepcją wzrokową.
Opisano różne zmiany związane z problemami z równowagą i koordynacją, które wynikają głównie z obecności nieprawidłowości układu mięśniowo-szkieletowego i które spowodują między innymi opóźnienie w nabywaniu chodu, końcową motorykę itp..
Możliwe jest stwierdzenie umiarkowanego upośledzenia umysłowego, typowe IQ osób dotkniętych chorobą oscyluje zwykle między 60 a 70. Jeśli chodzi o obszary dotknięte chorobą, istnieje wyraźna asymetria: oprócz koordynacji psychomotorycznej, percepcji i integracji wzrokowej zwykle być wyraźnie dotknięte, podczas gdy obszary takie jak język są zwykle bardziej rozwinięte.
Na najbardziej początkowych etapach zwykle występuje opóźnienie w nabywaniu umiejętności językowych, jednak zwykle ustępuje ono około 3-4 lat. Dzieci z zespołem Williamsa mają zwykle dobrą ekspresyjną komunikację, potrafią używać kontekstowego słownictwa, poprawnej gramatyki, kontaktu wzrokowego, mimiki itp..
Jednym z najbardziej znaczących ustaleń w zespole Williamsa jest wyjątkowe zachowanie społeczne osób dotkniętych chorobą. Chociaż w niektórych przypadkach mogą wystąpić kryzysy lękowe lub nadmierne zmartwienia, są one bardzo empatyczne i wrażliwe.
Najnowsze badania wykazały, że przyczyną zespołu Williamsa są różne zmiany genetyczne na chromosomie 7. Chromosomy przenoszą informację genetyczną każdej osoby i są zlokalizowane w jądrach komórek organizmu..
U ludzi możemy znaleźć 46 chromosomów, które są rozmieszczone parami. Są one ponumerowane od 1 do 23, z wyjątkiem ostatniej pary chromosomów płci, zwanej XX w przypadku kobiet, XY w przypadku mężczyzn. Zatem w każdym chromosomie może znajdować się nieskończona liczba genów.
W szczególności nieprawidłowym procesem zidentyfikowanym w zespole Williamsa jest mikroselekcja lub rozpad cząsteczki DNA, która potwierdza ten chromosom. Zwykle ten typ błędu ma miejsce w fazie rozwoju gamet męskich lub żeńskich..
Nieprawidłowości genetyczne występują na obszarze 7q11.23, w którym zidentyfikowano ponad 25 różnych genów związanych z charakterystycznym klinicznym wzorcem tej patologii..
Niektóre z genów, takie jak Clip2, ELN, GTF21, GTF2IRD1 lub LIMK1, są nieobecne u osób dotkniętych chorobą. Utrata ELN jest związana z nieprawidłowościami dotyczącymi tkanki łącznej, skóry i układu sercowo-naczyniowego.
Z drugiej strony, niektóre badania wskazują, że utrata genów Clip2, GTF2I, GTF2IRD1 i LIMK1 może wyjaśniać zmiany w procesach widzenia, fenotypie behawioralnym lub deficytach poznawczych.
Ponadto, w szczególności, gen GTF2IRD1 wydaje się odgrywać znaczącą rolę w rozwoju nietypowych rysów twarzy. Z kolei gen NCF1 wydaje się być powiązany z wysokim ryzykiem rozwoju nadciśnienia.
Do ostatnich lat rozpoznanie zespołu Williamsa było dokonywane wyłącznie na podstawie obserwacji cech fenotypowych (m.in. zmiany twarzy, niepełnosprawność intelektualna, określone deficyty poznawcze).
Jednak obecnie rozpoznanie zespołu Williamsa zazwyczaj przebiega w dwóch etapach: analiza wyników klinicznych i potwierdzające badania genetyczne. Tak więc diagnoza kliniczna zwykle obejmuje:
- Badanie i ocena fizyczna i neurologiczna.
- Analiza parametrów wzrostu.
- Badanie układu krążeniowo-oddechowego.
- Badanie nefrologiczne.
- Analiza poziomu wapnia w moczu i krwi.
- Analiza okulistyczna.
Z drugiej strony, analiza genetyczna służy do potwierdzenia obecności zmian genetycznych zgodnych z zespołem Williamsa, a do najczęściej stosowanych testów należy technika fluorescencyjnej hybrydyzacji in situ (FIHS)..
Po ekstrakcji próbki krwi przeprowadza się technikę hybrydyzacji in situ poprzez znakowanie sond DNA, które są wykrywane w świetle fluorescencyjnym..
Nie ma specyficznego leczenia zespołu Williamsa, jednak patologia ta wiąże się z wieloma powikłaniami w różnych narządach, więc interwencje medyczne będą ukierunkowane na leczenie tych narządów..
Autorzy González Fernández i Uyaguari Quezada podkreślają, że wszystkie interwencje muszą mieć wyraźny charakter multidyscyplinarny, pozwalający na leczenie objawowej różnorodności charakterystycznej dla tego zespołu. Ponadto wskazują również różne środki terapeutyczne w zależności od dotkniętego obszaru:
W takim przypadku powikłania medyczne, takie jak zmiany kardiologiczne lub wady rozwojowe układu mięśniowo-szkieletowego, wymagają zwykle leczenia opartego głównie na podawaniu leków i zabiegach chirurgicznych. W leczeniu objawów fizycznych zwykle uczestniczą lekarze z różnych dziedzin (pediatrzy, kardiolodzy, okuliści itp.)..
Deficyty poznawcze, takie jak zmiany wzrokowo-percepcyjne lub opóźnienia językowe, należy rozwiązywać na wczesnych etapach. Stymulacja poznawcza i rehabilitacja będą decydującym czynnikiem w osiągnięciu autonomicznego życia w wieku dorosłym.
Chociaż osoby dotknięte zespołem Williamsa zwykle prezentują dobre funkcjonowanie społeczne, w niektórych przypadkach mają tendencję do wykazywania nadmiernie lękowych zachowań i rozwijania zachowań wytrwałych lub fobii.
Dlatego w takich przypadkach konieczne będzie wdrożenie podejścia psychologicznego, poprzez różne strategie, które są skuteczne w celu zminimalizowania tych problemów lub trudności..


Jeszcze bez komentarzy