Plik priony są to białka bez genomu lub kwasów nukleinowych, które działają jak czynniki zakaźne. Termin „prion” oznacza białkową cząstkę zakaźną (z angielskiego Proteinaceous Infectious Particles) i został wymyślony przez neurologa i zdobywcę nagrody Nobla, Stanleya B. Prusinera.
W 1982 roku Prusiner i jego współpracownicy zidentyfikowali zakaźną cząsteczkę białka, badając przyczyny chorób Creutzfeldta-Jakoba (u ludzi) i gąbczastej encefalopatii bydła..
Te rzadkie czynniki zakaźne znajdują się w błonie prawidłowych komórek, jedynie jako białka o nieprawidłowym fałdowaniu i / lub o nieprawidłowej strukturze trójwymiarowej. Białka te są odpowiedzialne za wiele chorób zwyrodnieniowych i bardzo wysoką śmiertelność, które wpływają na tkanki nerwowe i strukturę mózgu..
Nazywa się je również chorobami prionowymi. Do najważniejszych, które dotykają ludzi, należą kuru, choroba Gerstmanna-Sträusslera-Scheinkera, zespół Creutzfeldta-Jakoba i śmiertelna bezsenność rodzinna..
Indeks artykułów
Priony to struktury białkowe obecne w błonach komórkowych. Te białka mają zmieniony kształt lub konformację [PrP (Sc)].
Jeśli chodzi o jej rozmnażanie, odbywa się to poprzez konwersję form, jak w przypadku choroby trzęsawki owiec. W tej chorobie priony rekrutują PrP (C) (białka prionowe o niezmienionej konformacji), aby stymulować konwersję do izoformy PrP (Sc)..
Powoduje to reakcję łańcuchową, która rozprzestrzenia materiał zakaźny, a tym samym umożliwia nawadnianie choroby. Nadal nie wiadomo, jak przebiega ten proces konwersji.
Te niezwykłe białka zdolne do namnażania nie mają kwasów nukleinowych. Dowodem na to jest to, że są odporne na promieniowanie rentgenowskie i promieniowanie ultrafioletowe. Te środki łatwo rozkładają kwasy nukleinowe.
Białka prionowe, z których składają się priony (PrP), występują w całym organizmie, nie tylko u ludzi, ale także u innych zdrowych kręgowców. Te białka są ogólnie odporne na proteazy (enzymy katalizujące białka).
Niewiele wiadomo na temat przydatności białek prionowych PrP (C), normalnej postaci niezakaźnego białka w organizmie człowieka..
Jednak niektórym badaczom udało się wykazać, że u myszy białka te aktywują naprawę mieliny w komórkach obwodowego układu nerwowego. Wykazano również, że ich brak powoduje demielinizację takich komórek nerwowych..
Wiedza, jaką posiadamy o budowie prionów, tkwi głównie w badaniach przeprowadzonych w bakterii Escherichia coli.
Badania wykazały, że łańcuchowe polipeptydy PrP (C) (normalne) i PrP (Sc) (zakaźne) są identyczne pod względem składu aminokwasów, ale różnią się konformacją 3D i fałdowaniem..
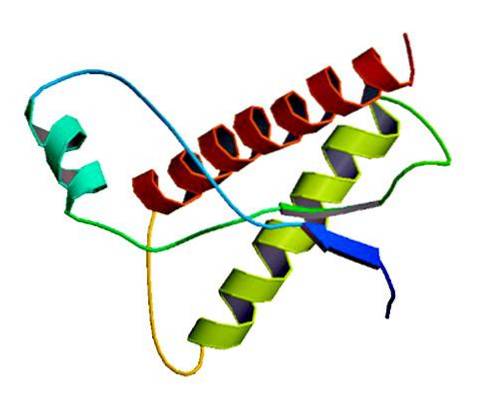
Te niezakaźne priony mają u ludzi 209 aminokwasów. Mają wiązanie dwusiarczkowe. Jego struktura jest alfa-helikalna, co oznacza, że ma aminokwasy w kształcie spirali (helisy alfa) i kilka płaskich pasm aminokwasów (arkusze beta)..
Białka tego nie można oddzielić przez wirowanie, co oznacza, że nie ulega sedymentacji. Jest łatwo trawiony przez proteazę serynową o szerokim spektrum działania zwaną proteinazą K..
Jest to zakaźne białko, które przekształca PrP (C) w zakaźne izoformy PrP (Sc) o nieprawidłowej konfiguracji lub kształcie.
Niewiele wiadomo na temat jego struktury 3D, jednak wiadomo, że ma kilka śrubowych kształtów i więcej płaskich pasm lub arkuszy beta. Przejście do izoformy jest tak zwanym kluczowym wydarzeniem w przypadku chorób prionowych.
Komórkowe białka prionowe [Prp (C)] są zlokalizowane na powierzchni komórek wielu różnych narządów i tkanek. Niewiele wiadomo na temat fizjologicznych funkcji prionów w organizmie. Mimo to eksperymenty przeprowadzone na myszach wskazują na możliwe funkcje, takie jak:
Wykazano, że PrP (C) działa z receptorami glutaminianu (jonotropowymi i metabotropowymi). PrP (C) uczestniczy jako receptor synaptotoksycznych oligomerów peptydu Aβ powierzchni komórki.
U myszy z rodziny Murinae odkryto, że białka prionowe PrP (C) ulegają ekspresji w ciągu kilku dni po implantacji, w rozwoju embrionalnym.
Wskazuje to, że odgrywają rolę w rozwoju tych małych ssaków. Rola, która zdaniem badaczy wiąże się z regulacją neurytogenezy (produkcja aksonów i dendrytów neuronów).
Działają również na wzrost aksonów. Te białka prionowe są nawet zaangażowane w rozwój obwodu móżdżku. Z tego powodu uważa się, że brak tych prionów PrP (C) pociąga za sobą opóźnienie w rozwoju motorycznym gryzoni..
W badaniach nad nadekspresją PrP (C) przez orientację genów stwierdzono, że brak tych prionów powoduje problemy z dopływem krwi do niektórych części mózgu (ostre niedokrwienie mózgu).
Oznacza to, że białka prionowe działają jako neuroprotektory. Ponadto wykazano, że nadekspresja PrP (C) może zmniejszyć lub poprawić urazy spowodowane niedokrwieniem..
Niedawno odkryto fizjologiczną rolę Prp (C) w utrzymaniu mieliny obwodowej.
Podczas badań laboratoryjnych odkryto, że pod nieobecność białka prionowego u myszy laboratoryjnych wystąpiły niedobory w nerwach przenoszących informacje z mózgu i rdzenia kręgowego, co nazywa się neuropatią obwodową..
Istnieją białka podobne do prionów, które znajdują się w innych częściach ciała niż mózg.
Funkcją takich białek jest inicjowanie, regulowanie i / lub kontrolowanie śmierci komórki, gdy organizm jest atakowany (na przykład przez wirony), zapobiegając w ten sposób rozprzestrzenianiu się patogenu..
Ta szczególna funkcja tych białek skłania badaczy do rozważenia możliwego znaczenia niezakaźnych prionów w walce z patogenami..
Badanie przeprowadzone w Stowers Institute w Missouri w USA wykazało, że priony PrP mogą odgrywać rolę w utrzymaniu pamięci długotrwałej.
Badanie wykazało, że niektóre białka prionowe mogą być kontrolowane w celu utrzymania fizjologicznych funkcji pamięci długotrwałej..
Badanie białek prionowych, które ulegają ekspresji w komórkach macierzystych tkanki krwi, wykazało, że wszystkie te komórki macierzyste (krwiotwórcze) wyrażają białka prionowe w błonie komórkowej. Za co uważa się, że uczestniczą w złożonym i bardzo ważnym procesie odnowy komórek.
Patologie pochodzenia prionowego są uznawane za postępujące zwyrodnieniowe zaburzenia mózgu. Mogą atakować bydło, jelenie, karibu, owce, a nawet ludzi.
Choroby te są spowodowane zmianą struktury białek PrP (C) i których specyficzne funkcje są nadal niepewne. Patologie prionowe mogą pojawić się bez znanej przyczyny. Mogą mieć odziedziczone pochodzenie genetyczne, a także mogą być przenoszone w sposób zakaźno-zaraźliwy.
Priony powodują choroby rodzinne, sporadyczne i zaraźliwe. Rodzinne choroby prionowe to te, które są dziedziczne. Sporadyczne patologie są najczęstsze i występują bez znanych przyczyn..
Choroby zakaźne są uważane za rzadkie, przenoszone są z człowieka na człowieka, ze zwierzęcia na zwierzę, z człowieka na zwierzę i odwrotnie. Przyczyny są wielorakie i sięgają od spożycia skażonego mięsa, kanibalizmu, transfuzji po manipulowanie skażonym sprzętem chirurgicznym..
Najczęstsze choroby prionowe to:
Uważana za najpowszechniejszą chorobę prionową wśród ludzi, jest chorobą kosmopolityczną, to znaczy rozprzestrzenia się na całym świecie. Może być dziedziczna (rodzinna), sporadyczna lub zakaźna.
Pacjenci zgłaszają się z objawami, takimi jak demencja, szarpnięcia lub nagłe mimowolne ruchy i niedobory ośrodkowego układu nerwowego.
W zależności od zastosowanego leczenia i postaci choroby, śmierć może nastąpić w okresie od 4 miesięcy do 2 lat po nabyciu choroby. Diagnoza jest trudna do postawienia, zwykle się ją robi post morten, podczas sekcji zwłok.
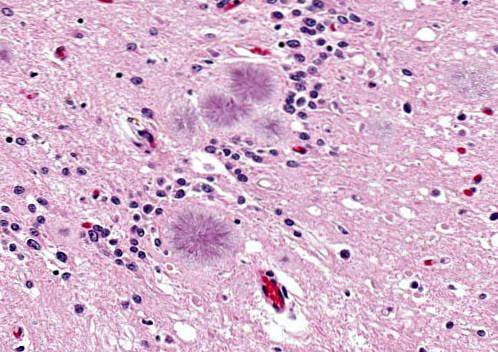
Jest to choroba wywoływana przez priony w dziedzicznym lub autosomalnie dominującym zakaźnym procesie mózgowym. Choroba objawia się u osób w wieku od 40 do 60 lat.
Osoby te wykazują problemy z wymawianiem słów (dyzartria), szarpnięcia lub nagłe mimowolne ruchy, często występuje agresywność.
Objawiają się zwyrodnieniem móżdżku, któremu towarzyszy chwiejny chód. Można również zaobserwować między innymi hiporefleksję, głuchotę, paraliż wzroku, demencję. Średnia długość życia wynosi około 5 lat lub trochę dłużej.
Jest to choroba bardzo rzadka, do tego stopnia, że zakres jej występowania wynosi od 2 do 3 przypadków na 100 milionów mieszkańców. Patologia jest podobna do choroby Gerstmanna-Sträusslera-Scheinkera.
Kliniczne objawy białka wskazują na niską oporność na proteazy, niektóre są bardziej, a inne mniej wrażliwe na te enzymy.
Objawy, które zgłaszają pacjenci to: problemy z mową i zaburzeniami poznawczymi, utrata neuronów w obszarze, w którym mózg kontroluje ruchy i koordynuje mięśnie.
Choroba występuje często u pacjentów w podeszłym wieku (70 lat), a szacowany czas życia po zakażeniu wynosi około 20 miesięcy.
Jest to choroba dziedziczna lub rodzinna, może również występować sporadycznie. Wiadomo, że choroba jest spowodowana dziedziczną lub autosomalną dominującą mutacją.
Pacjenci zgłaszają objawy, takie jak narastające problemy ze snem i utrzymaniem snu, demencja, zaburzenia poznawcze, a nawet problemy z nadciśnieniem, tachykardią, nadmierną potliwością i innymi..
Wiek, na który wpływa, jest dość szeroki i waha się od 23 do 73 lat, jednak średni wiek wynosi 40 lat. Żywotność po zakażeniu wynosi nieco ponad 6 lat.
Ta choroba prionowa została wykryta tylko u mieszkańców Papui-Nowej Gwinei. Jest to choroba związana z kanibalizmem i kulturową tradycją rytuału żałoby po zmarłych, gdzie ludzie ci jedzą mózg lub ludzkie mięso.
Osoby, które są nosicielami choroby, zwykle mają niekontrolowane i mimowolne ruchy w różnych częściach ciała.
Objawiają się drżeniem, utratą kontroli nad ruchami i utratą koordynacji mięśni. Oczekiwana długość życia u zarażonych osób wynosi dwa lata.
Wśród patologii wywoływanych przez priony u zwierząt jest gąbczasta encefalopatia bydła. Choroba ta spowodowała spustoszenie w Europie, w zdrowiu publicznym, zwierzętach oraz w gospodarce dotkniętych nią krajów.
Inne choroby zwierząt obejmują trzęsawkę, zakaźną encefalopatię norek, przewlekłą chorobę wyniszczającą (u jeleni) i gąbczastą encefalopatię kotów..
Chorobom tym, podobnie jak ludziom, brakuje skutecznego leczenia, dlatego profilaktyka jest niezbędna, zwłaszcza po zakażeniach u ludzi, które wystąpiły w wyniku spożycia mięsa zakażonych krów..
Do chwili obecnej nie jest znane lekarstwo na choroby prionowe. Leczenie jest objawowe. Pacjentom zaleca się planowanie opieki paliatywnej i badań genetycznych oraz doradztwo dla członków rodziny.
Na pacjentach z chorobami prionowymi testowano szeroką gamę leków, takich jak leki przeciwwirusowe, przeciwnowotworowe, leki na choroby, takie jak choroba Parkinsona, leki immunosupresyjne, antybiotyki, leki przeciwgrzybicze, a nawet leki przeciwdepresyjne..
Jednak obecnie nie ma dowodów wskazujących, że niektóre z nich zmniejszają objawy lub poprawiają przeżycie pacjentów..
Priony są odporne na różnorodne zmiany fizyczne i chemiczne. Jednak stosuje się różne techniki, aby uniknąć zakażenia pacjentów zanieczyszczonymi narzędziami chirurgicznymi..
Jedną z najczęściej stosowanych technik jest sterylizacja sprzętu w autoklawie w temperaturze 132 ° C przez jedną godzinę, a następnie zanurzenie instrumentów w wodorotlenku sodu na co najmniej jedną godzinę..
Z drugiej strony Światowa Organizacja Zdrowia (WHO) opracowała środki zapobiegające rozprzestrzenianiu się chorób prionowych. Organizacja ta ustala normy postępowania z zakazanymi lub potencjalnie ryzykownymi tkankami, takimi jak: oczy, mózg, jelita, migdałki i rdzeń kręgowy.


Jeszcze bez komentarzy