Plik ketogeneza Jest to proces, w którym otrzymuje się acetooctan, β-hydroksymaślan i aceton, które razem nazywane są ciałami ketonowymi. Ten złożony i precyzyjnie regulowany mechanizm zachodzi w mitochondriach w wyniku katabolizmu kwasów tłuszczowych.
Uzyskanie ciał ketonowych ma miejsce, gdy organizm poddawany jest wyczerpującym okresom postu. Chociaż metabolity te są w większości syntetyzowane w komórkach wątroby, są one ważnym źródłem energii w różnych tkankach, takich jak mięśnie szkieletowe oraz tkanki serca i mózgu..
Ss-hydroksymaślan i acetooctan to metabolity stosowane jako substraty w mięśniu sercowym i korze nerek. W mózgu ciała ketonowe stają się ważnymi źródłami energii, gdy organizm wyczerpał zapasy glukozy.
Indeks artykułów
Ketogeneza jest uważana za bardzo ważną funkcję fizjologiczną lub szlak metaboliczny. Generalnie mechanizm ten zachodzi w wątrobie, chociaż wykazano, że można go przeprowadzić w innych tkankach zdolnych do metabolizowania kwasów tłuszczowych.
Tworzenie ciał ketonowych jest głównym pochodnym metabolizmu acetylo-CoA. Ten metabolit jest pozyskiwany ze szlaku metabolicznego znanego jako β-oksydacja, czyli degradacja kwasów tłuszczowych.
Dostępność glukozy w tkankach, w których zachodzi β-oksydacja, determinuje metaboliczne losy acetylo-CoA. W szczególnych sytuacjach utlenione kwasy tłuszczowe są kierowane prawie w całości do syntezy ciał ketonowych..
Głównym ciałem ketonowym jest acetooctan lub kwas acetooctowy, który jest syntetyzowany głównie w komórkach wątroby. Inne cząsteczki tworzące ciała ketonowe pochodzą z acetooctanu.
Redukcja kwasu acetooctowego prowadzi do powstania drugiego ciała ketonowego, D-β-hydroksymaślanu. Aceton jest związkiem trudnym do degradacji i powstaje w wyniku samoistnej reakcji dekarboksylacji acetooctanu (więc nie wymaga interwencji żadnego enzymu), gdy występuje we krwi w dużych stężeniach.
Oznaczenie ciał ketonowych zostało ustalone konwencją, ponieważ ściśle mówiąc β-hydroksymaślan nie ma funkcji ketonowej. Te trzy cząsteczki są rozpuszczalne w wodzie, co ułatwia ich transport we krwi. Jego główną funkcją jest dostarczanie energii niektórym tkankom, takim jak mięsień szkieletowy i mięsień sercowy.
Enzymy biorące udział w tworzeniu ciał ketonowych znajdują się głównie w komórkach wątroby i nerek, co wyjaśnia, dlaczego te dwie lokalizacje są głównymi producentami tych metabolitów. Jego synteza zachodzi wyłącznie i wyłącznie w macierzy mitochondrialnej komórek.
Zsyntetyzowane cząsteczki przechodzą do krwiobiegu, docierając do tkanek, które ich potrzebują, gdzie są degradowane do acetylo-CoA..
Losy metaboliczne acetylo-CoA z β-oksydacji zależą od wymagań metabolicznych organizmu. To utlenia się do COdwa i HdwaLub poprzez cykl kwasu cytrynowego lub syntezę kwasów tłuszczowych, jeśli metabolizm lipidów i węglowodanów jest stabilny w organizmie.
Kiedy organizm potrzebuje wytworzenia węglowodanów, szczawiooctan jest używany do produkcji glukozy (glukoneogenezy) zamiast rozpoczynania cyklu kwasu cytrynowego. Dzieje się tak, jak wspomniano, gdy organizm ma pewną niezdolność do uzyskania glukozy, w przypadkach takich jak długotrwały post lub obecność cukrzycy.
Dzięki temu powstały w wyniku utleniania kwasów tłuszczowych acetylo-CoA wykorzystywany jest do produkcji ciał ketonowych..
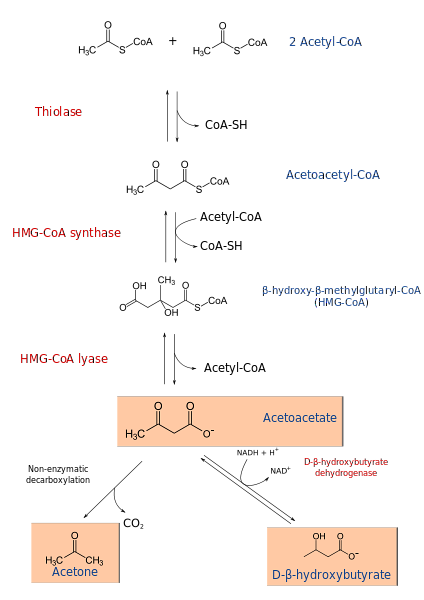
Proces ketogenezy rozpoczyna się od produktów β-utleniania: acetacetylo-CoA lub acetylo-CoA. Gdy substratem jest acetylo-CoA, pierwszy etap polega na kondensacji dwóch cząsteczek, reakcji katalizowanej przez transferazę acetylo-CoA, w celu wytworzenia acetacetylo-CoA.
Acetacetylo-CoA jest kondensowany z trzecim acetylo-CoA w wyniku działania syntazy HMG-CoA, z wytworzeniem HMG-CoA (β-hydroksy-β-metyloglutarylo-CoA). HMG-CoA ulega degradacji do acetooctanu i acetylo-CoA pod działaniem liazy HMG-CoA. W ten sposób uzyskuje się pierwsze ciało ketonowe.
Acetooctan jest redukowany do β-hydroksymaślanu przez interwencję dehydrogenazy β-hydroksymaślanowej. Ta reakcja zależy od NADH.
Głównym ciałem acetylooctanowym jest β-ketokwas, który ulega nieenzymatycznej dekarboksylacji. Ten proces jest prosty i wytwarza aceton i COdwa.
Ta seria reakcji prowadzi zatem do powstania ciał ketonowych. Będąc rozpuszczalnymi w wodzie, można je łatwo transportować przez krążenie krwi, bez konieczności mocowania do struktury albuminy, jak to ma miejsce w przypadku kwasów tłuszczowych, które są nierozpuszczalne w środowisku wodnym..
Metabolizm kwasów tłuszczowych wytwarza substraty do ketogenezy, więc te dwa szlaki są funkcjonalnie powiązane.
Acetoacetylo-CoA jest inhibitorem metabolizmu kwasów tłuszczowych, gdyż hamuje aktywność dehydrogenazy acylo-CoA, która jest pierwszym enzymem β-oksydacji. Ponadto działa hamująco na transferazę acetylo-CoA i syntazę HMG-CoA.
Enzym syntaza HMG-CoA, podporządkowany CPT-I (enzym zaangażowany w produkcję acylokarnityny w β-oksydacji), odgrywa ważną rolę regulacyjną w tworzeniu kwasów tłuszczowych.
Żywienie organizmów reguluje złożony zestaw sygnałów hormonalnych. Spożywane w pożywieniu węglowodany, aminokwasy i lipidy odkładają się w tkance tłuszczowej w postaci triacylogliceroli. Insulina, hormon anaboliczny, bierze udział w syntezie lipidów i tworzeniu triacylogliceroli.
Na poziomie mitochondrialnym β-oksydacja jest kontrolowana przez wnikanie i udział niektórych substratów w mitochondriach. Enzym CPT I syntetyzuje acylokarnitynę z cytozolowego Acylo-CoA.
Kiedy organizm jest karmiony, aktywowana jest karboksylaza acetylo-CoA, a cytrynian zwiększa poziom CPT I, podczas gdy jego fosforylacja spada (reakcja zależna od cyklicznego AMP).
Powoduje to gromadzenie się malonylo-CoA, który stymuluje syntezę kwasów tłuszczowych i blokuje ich utlenianie, zapobiegając powstaniu daremnego cyklu..
W przypadku postu aktywność karboksylazy jest bardzo niska, ponieważ poziomy enzymu CPT I zostały zmniejszone, a także został on fosforylowany, aktywując i promując utlenianie lipidów, co następnie pozwoli na tworzenie ciał ketonowych przez acetylo- CoA.
Ciała ketonowe dyfundują z komórek, w których zostały zsyntetyzowane, i są transportowane do tkanek obwodowych przez krwiobieg. W tych tkankach mogą być utleniane poprzez cykl kwasów trikarboksylowych.
W tkankach obwodowych β-hydroksymaślan jest utleniany do acetooctanu. Następnie obecny acetooctan jest aktywowany przez działanie enzymu transferazy 3-ketoacylo-CoA.
Sukcynylo-CoA działa jako dawca CoA, przekształcając się w bursztynian. Aktywacja acetooctanu ma na celu zapobieganie przekształcaniu się sukcynylo-CoA w bursztynian w cyklu kwasu cytrynowego, ze sprzężoną syntezą GTP przez działanie syntazy sukcynylo-CoA.
Powstały acetoacetylo-CoA ulega rozkładowi tiolitycznemu, tworząc dwie cząsteczki acetylo-CoA, które są włączane do cyklu kwasu trikarboksylowego, lepiej znanego jako cykl Krebsa..
W komórkach wątroby brakuje transferazy 3-ketoacylo-CoA, co zapobiega aktywacji tego metabolitu w tych komórkach. W ten sposób gwarantuje się, że ciała ketonowe nie utleniają się w komórkach, w których zostały wyprodukowane, ale mogą zostać przeniesione do tkanek, w których ich aktywność jest wymagana..
W organizmie człowieka wysokie stężenia ciał ketonowych we krwi mogą powodować szczególne stany zwane kwasicą i ketonemią..
Produkcja tych metabolitów odpowiada katabolizmowi kwasów tłuszczowych i węglowodanów. Jedną z najczęstszych przyczyn stanu patologicznej ketogenezy jest wysokie stężenie fragmentów diwęglanu octowego, które nie są degradowane na drodze utleniania kwasów trikarboksylowych..
W konsekwencji następuje wzrost poziomu ciał ketonowych we krwi powyżej 2 do 4 mg / 100 N i ich obecność w moczu. Skutkuje to zaburzeniem pośredniego metabolizmu tych metabolitów..
Przyczyną stanu hiperketonemii są pewne defekty przysadkowych czynników neurogruczołowych, które regulują degradację i syntezę ciał ketonowych, wraz z zaburzeniami metabolizmu węglowodorów..
Cukrzyca (typ 1) jest chorobą endokrynologiczną, która powoduje zwiększoną produkcję ciał ketonowych. Niewystarczająca produkcja insuliny uniemożliwia transport glukozy do mięśni, wątroby i tkanki tłuszczowej, gromadząc się w ten sposób we krwi.
Komórki pod nieobecność glukozy rozpoczynają proces glukoneogenezy i rozpadu tłuszczu i białka w celu przywrócenia ich metabolizmu. W konsekwencji stężenie szczawiooctanu spada, a utlenianie lipidów wzrasta..
Następuje wtedy nagromadzenie acetylo-CoA, które przy braku szczawiooctanu nie może podążać szlakiem kwasu cytrynowego, powodując tym samym wysoką produkcję ciał ketonowych, charakterystyczną dla tej choroby..
Nagromadzenie acetonu jest wykrywane na podstawie jego obecności w moczu i oddechu osób z tym schorzeniem i jest w rzeczywistości jednym z objawów wskazujących na manifestację tej choroby.



Jeszcze bez komentarzy