
Plik lizosomy Są organellami komórkowymi otoczonymi błonami znajdującymi się wewnątrz komórek zwierzęcych. Są to przedziały o kwaśnym pH i bogate w enzymy trawienne, zdolne do degradacji wszelkiego rodzaju cząsteczek biologicznych: białek, węglowodanów i kwasów nukleinowych.
Ponadto mogą degradować materiał z zewnątrz komórki. Z tego powodu lizosomy pełnią wiele funkcji w metabolizmie komórkowym, a dzięki bogatemu w enzymy hydrolityczne składowi często nazywane są „żołądkiem” komórki..
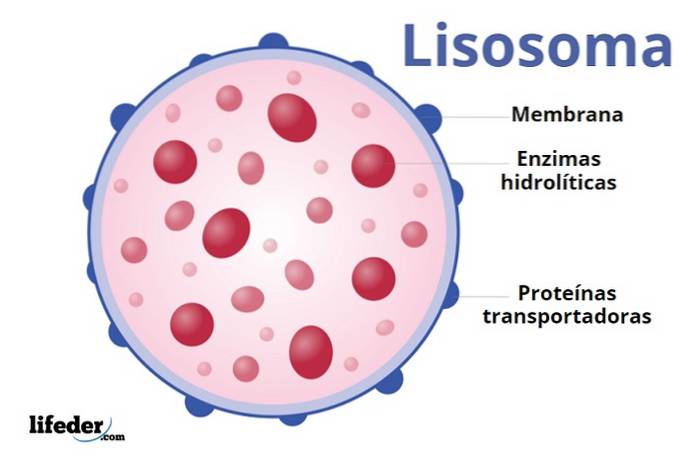
Lizosomy powstają w wyniku połączenia pęcherzyków, które wyłaniają się z aparatu Golgiego. Komórka rozpoznaje pewne sekwencje, które działają jako „znaczniki” enzymów hydrolitycznych i wysyła je do tworzących się lizosomów.
Te wakuole mają kulisty kształt, a ich rozmiar znacznie się różni, będąc dość dynamiczną strukturą komórkową..
Indeks artykułów
Lizosomy zostały odkryte ponad 50 lat temu przez badacza Christiana de Duve. Zespół De Duve przeprowadzał eksperymenty z wykorzystaniem techniki frakcjonowania subkomórkowego w celu zbadania lokalizacji niektórych enzymów..
Ten protokół eksperymentalny pozwolił na odkrycie organelli, ponieważ naukowcy zauważyli, że uwalnianie enzymów hydrolitycznych wzrosło w miarę dodawania związków niszczących błony..
Później udoskonalenie technik biologii molekularnej i istnienie lepszego sprzętu - takiego jak mikroskopy elektronowe - potwierdziły jego obecność. W rzeczywistości można by wywnioskować, że lizosomy zajmują 5% objętości wewnątrzkomórkowej.
Jakiś czas po jego odkryciu wykazano obecność enzymów hydrolitycznych w jego wnętrzu, przekształcając lizosom w swego rodzaju centrum degradacji. Ponadto lizosomy były związane z życiem endocytarnym.
Historycznie rzecz biorąc, lizosomy uważano za punkt końcowy endocytozy, wykorzystywano jedynie do degradacji cząsteczek. Obecnie wiadomo, że lizosomy są dynamicznymi przedziałami komórkowymi, zdolnymi do fuzji z różnymi dodatkowymi organellami..
Lizosomy to wyjątkowe przedziały komórek zwierzęcych, w których znajdują się różne enzymy zdolne do hydrolizy białek i trawienia niektórych cząsteczek..
Są gęstymi, kulistymi wakuolami. Rozmiar konstrukcji jest bardzo zróżnicowany i zależy od materiału, który został wcześniej uchwycony.
Lizosomy wraz z retikulum endoplazmatycznym i aparatem Golgiego są częścią układu endomembranowego komórki. Chociaż te trzy struktury są sieciami membranowymi, nie są one ze sobą ciągłe..
Główną cechą lizosomów jest znajdująca się w nich bateria enzymów hydrolitycznych. Istnieje około 50 enzymów zdolnych do degradacji szerokiego zakresu biocząsteczek.
Należą do nich nukleazy, proteazy i fosfatazy (które usuwają grupy fosforanowe z mononukleotydów fosfolipidowych i innych związków). Ponadto zawierają inne enzymy odpowiedzialne za rozkład polisacharydów i lipidów.
Logicznie rzecz biorąc, te enzymy trawienne muszą być przestrzennie oddzielone od reszty składników komórkowych, aby uniknąć ich niekontrolowanej degradacji. W ten sposób komórka może „wybrać” związki, które muszą zostać wyeliminowane, ponieważ może regulować elementy wchodzące do lizosomu.
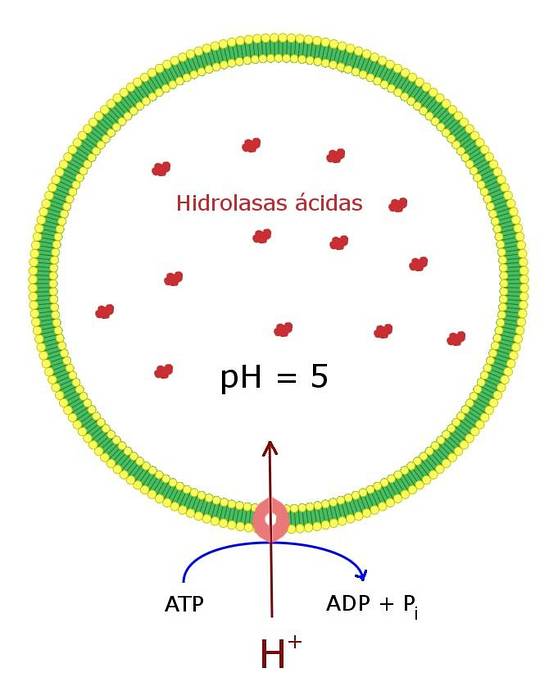
Wnętrze lizosomów jest kwaśne (blisko 4,8), a zawarte w nim enzymy dobrze działają w tych warunkach pH. Dlatego są znane jako kwaśne hydrolazy..
Kwaśne pH charakterystyczne dla tego przedziału komórkowego jest utrzymywane dzięki obecności pompy protonowej i kanału chlorkowego w membranie. Razem transportują kwas solny (HCl) do lizosomu. Pompa jest zakotwiczona w błonie organelli.
Zadaniem tego kwaśnego pH jest aktywacja różnych enzymów hydrolitycznych obecnych w lizosomie i unikanie - w miarę możliwości - ich aktywności enzymatycznej przy obojętnym pH cytozolu..
W ten sposób mamy już dwie bariery, które działają jako ochrona przed niekontrolowaną hydrolizą: utrzymywanie enzymów w izolowanym przedziale i że enzymy te działają dobrze przy kwaśnym pH tego przedziału..
Nawet w przypadku pęknięcia błony lizosomu uwolnienie enzymów nie miałoby większego wpływu - ze względu na obojętne pH cytozolu.
W składzie wewnętrznym lizosomu dominują enzymy hydrolityczne, dlatego są one ważnym regionem metabolizmu komórkowego, w którym następuje trawienie białek zewnątrzkomórkowych, które dostają się do komórki na drodze endocytozy, recykling organelli i białek cytozolowych..
Poniżej omówimy dogłębnie najważniejsze funkcje lizosomów: degradację cząsteczek przez autofagię i degradację przez fagocytozę..
Jeden mechanizm, który potrafi wychwytywać białka komórkowe, nazywa się autofagią „samożerności”. To zdarzenie pomaga utrzymać homeostazę komórkową, degradując struktury komórkowe, które nie są już potrzebne i przyczynia się do recyklingu organelli..
W wyniku tego zjawiska dochodzi do tworzenia się pęcherzyków zwanych autofagosomami. Są to małe obszary cytoplazmy lub innych przedziałów komórkowych, pochodzące z retikulum endoplazmatycznego, które łączą się z lizosomami.
Obie organelle mają zdolność łączenia się, ponieważ są ograniczone błoną plazmatyczną o charakterze lipidowym. Jest to analogiczne do próby dopasowania dwóch baniek mydlanych - utworzysz większą..
Po fuzji enzymatyczna zawartość lizosomu jest odpowiedzialna za degradację składników znajdujących się wewnątrz innego utworzonego pęcherzyka. Wychwytywanie tych cząsteczek wydaje się być procesem pozbawionym selektywności, powodując degradację białek znajdujących się w długo żyjącym cytozolu.
Wydaje się, że w komórce zdarzenie autofagii jest regulowane ilością dostępnych składników odżywczych.
Kiedy organizm odczuwa niedobór składników odżywczych lub doświadcza długotrwałego postu, aktywują się szlaki degradacji. W ten sposób komórka rozkłada białka, które nie są niezbędne, i uzyskuje możliwość ponownego wykorzystania niektórych organelli.
Świadomość, że lizosomy odgrywają ważną rolę w okresach postu, zwiększyła zainteresowanie naukowców tą organellą.
Oprócz aktywnego udziału w okresach niskiej wartości odżywczej lizosomy odgrywają ważną rolę w rozwoju niektórych linii istot organicznych..
W niektórych przypadkach rozwój pociąga za sobą całkowitą przebudowę organizmu, co oznacza, że niektóre narządy lub struktury muszą zostać wyeliminowane podczas tego procesu. Na przykład w metamorfozie owadów zawartość hydrolityczna lizosomów przyczynia się do przebudowy tkanek.
Endocytoza i fagocytoza odgrywają rolę w pobieraniu elementów zewnętrznych w stosunku do komórek i ich późniejszej degradacji.
Podczas fagocytozy niektóre komórki - takie jak makrofagi - są odpowiedzialne za połykanie lub rozkład dużych cząstek, takich jak bakterie lub szczątki komórek.
Wspomniane cząsteczki są wchłaniane przez wakuolę fagocytarną, zwaną fagosomem, która, podobnie jak w poprzednim przypadku, połączy się z lizosomami. Fuzja powoduje uwolnienie enzymów trawiennych wewnątrz fagosomu i cząsteczki ulegają degradacji.
Niektórzy autorzy wyróżniają ten przedział na dwa główne typy: typ I i typ II. Lizosomy typu I lub pierwotne są zaangażowane w przechowywanie enzymów hydrolitycznych, podczas gdy lizosomy wtórne są związane z procesami katalizy.
Tworzenie lizosomów rozpoczyna się od pobrania cząsteczek z zewnątrz przez pęcherzyki endocytarne. Te ostatnie łączą się z innymi strukturami zwanymi wczesnymi endosomami..
Później wczesne endosomy przechodzą proces dojrzewania, co prowadzi do powstania późnych endosomów.
W procesie formowania pojawia się trzeci składnik: pęcherzyki transportowe. Zawierają one hydrolazy kwasowe z sieci trans aparatu Golgiego. Obie struktury - pęcherzyki transportowe i późne endosomy - łączą się i stają się lizosomami, po uzyskaniu zestawu enzymów lizosomalnych.
W trakcie tego procesu recykling receptorów membranowych następuje poprzez recykling endosomów..
Kwaśne hydrolazy są oddzielane od receptora mannozy-6-fosforanu podczas procesu fuzji organelli, z których powstają lizosomy. Te receptory ponownie wchodzą do sieci trans Golgiego.
Powszechne jest zamieszanie między terminami endosomy i lizosom. Te pierwsze to przedziały komórkowe związane z błoną - jak lizosomy. Jednak kluczową różnicą między tymi dwoma organellami jest to, że lizosomy nie mają receptorów mannozo-6-fosforanu..
Oprócz tych dwóch bytów biologicznych istnieją inne typy pęcherzyków. Jedną z nich są wakuole, w których występuje głównie woda.
Pęcherzyki transportowe, jak sama nazwa wskazuje, uczestniczą w przemieszczaniu substancji do innych miejsc w komórce. W międzyczasie pęcherzyki wydzielnicze usuwają odpady lub substancje chemiczne (takie jak te zaangażowane w synapsę neuronów).
U ludzi mutacje genów kodujących enzymy lizosomalne są związane z ponad 30 chorobami wrodzonymi. Te patologie są objęte terminem „lizosomalne choroby spichrzeniowe”.
Co zaskakujące, wiele z tych stanów wynika z uszkodzenia pojedynczego enzymu lizosomalnego..
U osób dotkniętych chorobą konsekwencją posiadania niefunkcjonalnego enzymu wewnątrz lizosomów jest nagromadzenie się produktów przemiany materii.
Najczęstsza zmiana w osadzaniu lizosomów jest znana jako choroba Gauchera i jest związana z mutacją w genie, który koduje enzym odpowiedzialny za glikolipidy. Co ciekawe, choroba ta występuje dość często wśród ludności żydowskiej, dotykając 1 na 2500 osób..


Jeszcze bez komentarzy