
Plik Zespół Aperta lub akrocefalosyndaktylia typu I (ACS1) jest patologią pochodzenia genetycznego, która charakteryzuje się obecnością różnych zmian i wad rozwojowych w czaszce, twarzy i kończynach.
Na poziomie klinicznym zespół Aperta charakteryzuje się obecnością lub rozwojem spiczastej lub wydłużonej czaszki, zapadniętej okolicy twarzy ze zmianą w projekcji zębów, zrostem i zamknięciem kości i stawów palców, zmiennym upośledzeniem umysłowym, zaburzeniami mowy itp..
Chociaż ta patologia może być dziedziczna, w większości przypadków zespół Aperta występuje bez obecności wywiadu rodzinnego, głównie z powodu mutacji de novo w fazie ciąży..
Mechanizmy genetyczne powodujące zespół Aperta nie są dokładnie znane. Obecnie zidentyfikowano różne zmiany genetyczne, które są zdolne do wywołania tej patologii, zasadniczo związane z mutacjami w genie FGFR2.
Z drugiej strony rozpoznanie zespołu Aperta zwykle rozpoczyna się od klinicznego podejrzenia w okresie prenatalnym po wykryciu nieprawidłowości w rutynowych badaniach ultrasonograficznych i jest potwierdzane przeprowadzeniem badania genetycznego..
Jeśli chodzi o leczenie, nie ma rodzaju interwencji leczniczej w przypadku zespołu Aperta. Jednak w całej historii tej patologii opracowano różne specyficzne interwencje, które zwykle obejmują między innymi neurochirurgię, chirurgię twarzoczaszki, chirurgię szczękowo-twarzową, farmakoterapię, fizjoterapię, interwencję psychologiczną i neuropsychologiczną..
Indeks artykułów
Zespół Aperta to patologia genetyczna charakteryzująca się występowaniem różnych wad rozwojowych szkieletu na poziomie czaszki, twarzy i / lub kończyn.
Istotną zmianą zespołu Aperta jest przedwczesne lub wczesne zamknięcie szczelin czaszkowych, co powoduje nieprawidłowy wzrost pozostałych struktur twarzy i czaszki. Oprócz tego mogą również pojawić się wady rozwojowe kończyn górnych i dolnych, takie jak zespolenie palców rąk i nóg..
Z drugiej strony, może to również wpływać na zdolności poznawcze osób cierpiących na zespół Aperta, ze zmiennym nasileniem od łagodnego do umiarkowanego..
Chociaż Baumgartner (1842) i Wheaton (1894) poczynili pierwsze wzmianki o tym schorzeniu, dopiero w 1906 roku francuski lekarz specjalista Eugene Apert dokładnie opisał ten zespół i opublikował pierwszy raport kliniczny..
W swojej publikacji Eugene Apert opisuje zbiór nowych przypadków pacjentów dotkniętych dobrze zdefiniowanym wzorcem wad rozwojowych i charakteryzujących się charakterystycznymi oznakami i objawami tej patologii..
Tak więc dopiero w 1995 roku zidentyfikowano etiologiczne czynniki genetyczne zespołu Aperta. W szczególności Wilkie i wsp. Opisali obecność dwóch mutacji w genie FGFR2 u około 40 chorych pacjentów..
Ponadto zespół Aperta jest schorzeniem zaliczanym do chorób lub patologii charakteryzujących się kraniosynostozą (przedwczesne zamknięcie szwów czaszkowych).
Inne patologie należące do tej grupy to zespół Pfeiffera, zespół Crouzona, zespół Saethre-Chotzcena i zespół Carpentera..
Zespół aperta jest uważany za rzadką lub rzadką patologię, to znaczy ma częstość występowania mniej niż jeden przypadek na 15 000 mieszkańców populacji ogólnej..
W szczególności zespół Aperta występuje u jednej osoby na każde 160 000-200 000 urodzeń, a ponadto istnieje 50% prawdopodobieństwo przeniesienia tej patologii na poziomie dziedzicznym.
Ponadto, jeśli chodzi o dystrybucję według płci, nie zidentyfikowano większej częstości występowania u mężczyzn i kobiet, ani nie powiązano jej z określonymi grupami etnicznymi lub lokalizacjami geograficznymi..
Obecnie, biorąc pod uwagę, że zespół Aperta został zidentyfikowany około 1984 roku, w raportach klinicznych i literaturze medycznej, które opublikowały ponad 300 przypadków tej patologii.
Objawy kliniczne zespołu Aperta zwykle obejmują wady rozwojowe lub niepełny rozwój struktury czaszki, nietypowy fenotyp lub wzór twarzy oraz zmiany kostne kończyn..
W przypadku zespołu Aperta centralne zajęcie wiąże się z utworzeniem i zamknięciem struktury kostnej czaszki. Podczas rozwoju embrionalnego zachodzi proces zwany kreneosynostozą, charakteryzujący się przedwczesnym zamknięciem szwów czaszkowych.
Szczeliny lub szwy czaszkowe to rodzaj pasm tkanki włóknistej, których podstawowym celem jest połączenie kości tworzących czaszkę (czołowej, potylicznej, ciemieniowej i skroniowej).
W fazie ciąży i we wczesnym okresie poporodowym struktura kości czaszki jest utrzymywana razem dzięki tym włóknistym i elastycznym tkankom.
Zwykle kości czaszki zrastają się dopiero około 12 do 18 miesięcy. Obecność miękkich plam lub przestrzeni między kościami czaszki jest częścią normalnego rozwoju dziecka.
Dlatego przez cały okres dzieciństwa te szwy lub elastyczne obszary pozwalają mózgowi rosnąć w sposób przyspieszony, a dodatkowo chronią go przed uderzeniami.
Tak więc w zespole Aperta przedwczesne zamknięcie tych szwów czaszkowych i kości czaszki uniemożliwia prawidłowy rozwój czaszki i mózgu..
W związku z tym najczęstsze oznaki i objawy zespołu Aperta mogą obejmować:
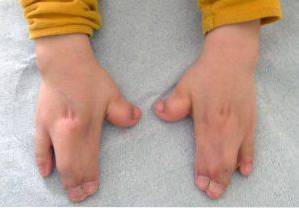
Tego typu zmiany dotyczą głównie kończyn górnych i dolnych, zwykle zrostu i rozwoju palców..
Oprócz tego można również zaobserwować inne objawy kliniczne na poziomie mięśniowo-szkieletowym, skrócenie różnych kości (promień, kość ramienna, kość udowa), niedorozwój łopatki lub miednicy, zespolenie kręgów szyjnych.
W konsekwencji wiele osób dotkniętych chorobą będzie miało ograniczoną ruchomość stawów, a zatem mogą mieć różne trudności w nabywaniu umiejętności motorycznych dużej i małej..
Te typy anomalii są bardzo niejednorodne i zmienne wśród osób dotkniętych chorobą, jednak zidentyfikowano niektóre z najczęstszych:
Etiologiczna zmiana tej patologii może prowadzić do rozwoju zmian chorobowych lub patologii wtórnych na poziomie morfologicznym i strukturalnym w różnych obszarach ciała, niektóre z nich obejmują:
Mimo, że w wielu przypadkach można zaobserwować ogólną zmianę funkcji poznawczych i poziomu intelektualnego, upośledzenie umysłowe nie występuje jednoznacznie we wszystkich przypadkach zespołu Aperta..
Ponadto w przypadkach, gdy występuje upośledzenie poziomu intelektualnego, może to być zmienne, w skali od łagodnego do umiarkowanego.
Z drugiej strony w obszarze językowym często dochodzi do rozwoju różnych deficytów, związanych głównie z artykulacją dźwięków w wyniku wad rozwojowych żuchwy i jamy ustnej..
Zespół Aperta wynika z obecności specyficznej mutacji w genie FGFR2. Badania eksperymentalne wykazały, że gen ten jest odpowiedzialny za produkcję białka zwanego receptorem czynnika wzrostu fibroblastów 2.
Wśród funkcji tego czynnika opisuje wysyłanie różnych sygnałów chemicznych do niedojrzałych komórek, aby spowodować ich transformację i różnicowanie w komórki kostne w fazie rozwoju płodowego lub prenatalnego..
Dlatego obecność mutacji w genie FGFR2 zmienia funkcjonowanie tego białka, a tym samym może powodować wczesną fuzję kości czaszki, dłoni i stóp..
Znaczną część klinicznych cech zespołu Aperta można zidentyfikować w czasie ciąży, szczególnie w badaniach ultrasonograficznych ciąży i rozwoju płodu..
Tak więc, gdy istnieje kliniczne podejrzenie, wznawia się badanie genetyczne w celu zidentyfikowania obecności mutacji genetycznej zgodnej z zespołem Aperta..
Z drugiej strony, gdy objawy są subtelne lub nie zostały zidentyfikowane przed urodzeniem, po tym można przeprowadzić szczegółową analizę fizyczną i różne testy genetyczne w celu potwierdzenia diagnozy..
Chociaż nie ma konkretnego lekarstwa na zespół Aperta, opisano różne podejścia do leczenia objawów i powikłań medycznych tej patologii..
Najskuteczniejsze interwencje terapeutyczne to takie, które są wdrażane wcześnie, w pierwszych chwilach życia i angażują specjalistów z różnych dziedzin.
Zazwyczaj leczenie chorych dzieci wymaga zindywidualizowanego planowania i zaplanowania wielu operacji. Dlatego leczenie tej patologii opiera się na korekcji wad rozwojowych układu kostnego i czaszkowo-twarzowego oraz wsparciu psychologicznym i neuropsychologicznym..
Celem neurochirurgii jest rekonstrukcja sklepienia czaszki, a specjaliści chirurgii szczękowo-twarzowej starają się korygować wady rozwojowe twarzy. Z drugiej strony częsty jest też udział chirurgów urazowych w rekonstrukcji wad rozwojowych dłoni i stóp.
Ponadto zaprojektowanie zindywidualizowanych programów wczesnej stymulacji, rehabilitacji komunikacyjnej, treningu umiejętności społecznych lub kontynuacji psychopedagogicznej jest korzystne dla osiągnięcia optymalnego, funkcjonalnego i niezależnego rozwoju osób dotkniętych chorobą..


Jeszcze bez komentarzy