
Plik Zespół MELAS Jest to rodzaj choroby mitochondrialnej pochodzenia dziedzicznego, która charakteryzuje się powodowanymi przez nią zaburzeniami neurologicznymi. Ta patologia jest zasadniczo definiowana przez prezentację encefalopatii mitochondrialnej, kwasicy mleczanowej i epizodów podobnych do udarów..
Na poziomie klinicznym objawy przedmiotowe i podmiotowe zespołu MELAS są zwykle widoczne przed 40. rokiem życia i są związane między innymi z napadami drgawkowymi, zaburzeniami świadomości lub incydentami naczyniowo-mózgowymi..
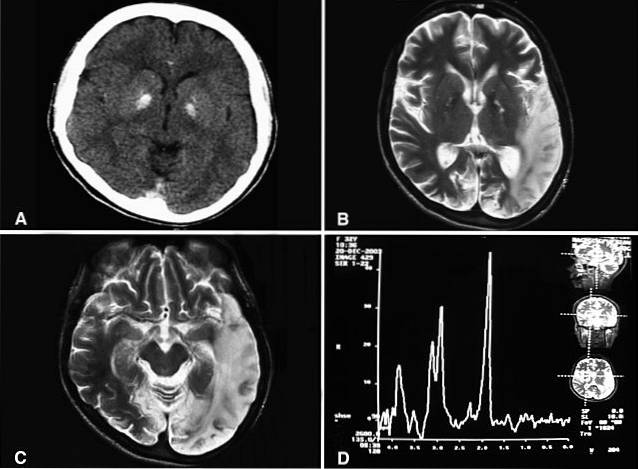
Ta patologia ma genetyczne pochodzenie etiologiczne związane ze specyficznymi mutacjami w mitochondrialnym DNA i nieprawidłowościami w łańcuchach enzymatycznych. W przypadku podejrzenia klinicznego rozpoznanie zespołu MELAS obejmuje zwykle różne badania laboratoryjne, takie jak elektroencefalografia (EEG), komputerowa tomografia osiowa czaszki (CT), rezonans magnetyczny (MRI) i badanie genetyczne.
Nie ma lekarstwa na zespół MELAS. Podejścia terapeutyczne koncentrują się na kontroli objawów i opiece paliatywnej. Biorąc pod uwagę zwyrodnieniowy i przewlekły charakter choroby MELAS, rokowanie medyczne wiąże się z poważnymi powikłaniami (zaburzenia sercowo-płucne, nerek, metaboliczne i neurologiczne).
Indeks artykułów
Zespół MELAS został po raz pierwszy opisany przez Shapiro i jego grupę roboczą w 1975 r. Jednak to Pavlakis (1984) użył nazwy MELAS jako akronimu dla jego najbardziej charakterystycznych przejawów..
W swoim raporcie klinicznym Pavlakis odniósł się do przebiegu klinicznego charakteryzującego się połączeniem napadów padaczkowych, postępującego upośledzenia mowy, kwasicy mleczanowej i zerwania czerwonych włókien mięśniowych..
To Pavlakis i Hirado ustalili kliniczne kryteria zespołu MELAS: napady padaczkowe, demencję, kwasicę mleczanową, postrzępione czerwone włókna i epizody podobne do udarów przed 40 rokiem życia..
Prezentacja tego zespołu jest bardzo zróżnicowana, a jego przebieg kliniczny jest zwykle widoczny przed czwartą dekadą życia. Rokowanie medyczne jest zwykle złe, a osoby dotknięte chorobą postępują z poważnymi komplikacjami medycznymi aż do śmierci.
Zespół MELAS to rzadka choroba, która zwykle rozpoczyna się w dzieciństwie lub w okresie dojrzewania, zwykle między 2 a 15 rokiem życia. Szczególnie wpływa na układ nerwowy i budowę mięśniową organizmu.
Niektóre z jego cech klinicznych obejmują drgawki, nawracające bóle głowy, wymioty, utratę apetytu, epizody podobne do udarów, zaburzenia świadomości, zaburzenia widzenia i słuchu oraz inne rodzaje zaburzeń motorycznych i poznawczych..
Zespół ten zawdzięcza swoją nazwę kardynalnym cechom klinicznym, które go definiują: encefalomiopatia mitochondrialna (encefalompyopatia mitochondrialna) ja; kwasica mleczanowa (kwasica mleczanowa) THE; epizody podobne do udarów S (genetyka).
Zespół MELAS jest często klasyfikowany jako choroba mitochondrialna lub mitochondrialna encefalomiopatia.
Choroby mitochondrialne stanowią szeroką grupę patologii charakteryzujących się obecnością zmian neurologicznych pochodzenia dziedzicznego spowodowanych określonymi mutacjami w DNA jądrowym lub mitochondrialnym.
Mitochondrium to rodzaj organelli komórkowych znajdujących się w cytoplazmie. Te organelle są niezbędne do metabolizmu energetycznego komórek naszego ciała. Odpowiada za pozyskiwanie energii z procesu utleniania do produkcji ATP. Ponadto składnik ten ma swoje własne wyposażenie genetyczne, mitochondrialne DNA..
Proces produkcji energii obejmuje wiele różnych mechanizmów biochemicznych, a częstą anomalią w chorobach mitochondriów jest zmiana końcowej fazy mechanizmu oksydacyjnego..
To mitochondrialny łańcuch oddechowy, który powoduje znaczny spadek produkcji energii w postaci ATP. Z tego powodu choroby mitochondrialne mogą objawiać się poważnymi nieprawidłowościami wieloukładowymi, w tym zmianami neurologicznymi i naczyniowo-mózgowymi.
Najczęstsze to zespół MERRF, zespół Kearnsa-Sayre'a i zespół MELAS..
Zespół MELAS jest rzadką chorobą w populacji ogólnej. Chociaż jego specyficzna częstość występowania nie jest dokładnie znana, jest to jedno z najczęstszych zaburzeń klasyfikowanych w ramach chorób mitochondrialnych..
Na całym świecie choroby mitochondrialne występują u około 1 przypadku na 4000 osób na całym świecie.
Jeśli chodzi o cechy socjodemograficzne, na poziomie międzynarodowym nie stwierdzono upodobań do jakiejkolwiek płci, grupy etnicznej / rasowej ani określonego pochodzenia geograficznego..
Zespół MELAS definiuje się na podstawie trzech głównych objawów klinicznych: encefalopatii mitochondrialnej, kwasicy mleczanowej i epizodów podobnych do udaru..
Encefalopatia to termin, który jest zwykle używany do określania tych zaburzeń lub patologii, których niejednorodny przebieg kliniczny ma swoje źródło w strukturalnych i funkcjonalnych nieprawidłowościach ośrodkowego układu nerwowego.
Na poziomie neurologicznym zespół MELAS charakteryzuje się nawracającymi napadami drgawkowymi. Napady są definiowane przez rozwój przejściowych epizodów nadmiernego pobudzenia motorycznego, obecność gwałtownych i mimowolnych ruchów mięśni, postrzeganie nieprawidłowych wrażeń lub zmienionej świadomości.
Napady mogą mieć zróżnicowany przebieg, być ogniskowe lub uogólnione:
Kliniczne nasilenie napadów polega na ich potencjalnej zdolności do trwałego uszkodzenia struktur nerwowych, co prowadzi do następstw poznawczych i psychomotorycznych.
Ze względu na nieprawidłowości w mechanizmach oksydacyjnych zaangażowanych w produkcję energii w organizmie zespół MELAS zwykle wiąże się z nieprawidłowym i patologicznym nagromadzeniem kwasu mlekowego..
Kwas mlekowy to substancja biochemiczna, która powstaje w wyniku rozpadu węglowodanów, gdy wykorzystujemy je jako formę energii przy niskim poziomie tlenu (niewydolność oddechowa, wysiłek fizyczny itp.).
Substancja ta jest zwykle wytwarzana głównie w krwinkach czerwonych i komórkach mięśniowych. W normalnych warunkach kwas mlekowy jest usuwany z organizmu przez wątrobę. Jednak obecność nienormalnie wysokich poziomów prowadzi do rozwoju kwasicy..
Kwasica zwykle generuje anomalie medyczne o dużym znaczeniu, które mogą prowadzić do śmierci chorego.
Niektóre z charakterystycznych objawów tego stanu to nudności, wymioty, biegunka, letarg, ból żołądka, bardzo zmieniony poziom świadomości, zaburzenia oddychania, niedociśnienie tętnicze, odwodnienie, a nawet wstrząs medyczny..
Epizody podobne do udaru charakteryzują się tym, że są podobne do udarów mózgu lub udaru mózgu. Zdarzenia te charakteryzują się obecnością ogniskowych zmian neurologicznych, spontanicznym pojawieniem się i ograniczonym czasem trwania..
Mają tendencję do preferencyjnego wpływu na okolice potyliczne, powodując zaburzenia widzenia. Jednak także jej częste nieprawidłowości językowe, sensoryczne czy motoryczne.
Identyfikacja wielu procesów wielozawałowych w różnych obszarach mózgu powoduje cierpienie związane z postępującym pogorszeniem funkcji poznawczych, prowadzącym do demencji.
Obecność opisanych powyżej cech klinicznych prowadzi do rozwoju różnych wtórnych objawów przedmiotowych i podmiotowych. Chociaż przebieg kliniczny zespołu MELAS może być bardzo niejednorodny, najczęściej obserwuje się niektóre z następujących cech:
Oprócz tych wyników, objawy psychiatryczne są również często powszechne w zespole MELAS. Niektóre z najczęstszych to:
W innych przypadkach można wyróżnić inne warunki, takie jak:
Zespół MELAS jest spowodowany obecnością zmian w mitochondrialnym DNA. Tego typu anomalie są dziedziczone po matce, ponieważ ten typ DNA w przypadku ojca jest tracony podczas zapłodnienia.
Na poziomie genetycznym pochodzenie zespołu MELAS powiązano z określonymi mutacjami w różnych genach: MT-TV, MT-TL1, MT-TH, MT-ND5, MT-ND1. Ten zestaw genów jest zwykle zlokalizowany w materiale genetycznym (DNA) mitochondriów komórkowych.
Wiele z tych genów odgrywa istotną rolę w produkcji białek biorących udział w przemianie cukrów, tłuszczów i tlenu w energię. Jednak inne pośredniczą w produkcji cząsteczek tRNA niezbędnych w budowaniu struktury aminokwasów..
W rozpoznaniu zespołu MELAS konieczne jest zidentyfikowanie wysokiego wskaźnika podejrzenia klinicznego, czyli ocena wszystkich cech klinicznych osoby dotkniętej chorobą. W każdym razie badanie historii medycznej indywidualnej i matczynej jest bardzo istotne..
Aby potwierdzić diagnozę i wykluczyć inne patologie, konieczne jest wykonanie różnych testów uzupełniających:
Obecnie nie ma lekarstwa na zespół MELAS..
Stosowanie procedur eksperymentalnych (podanie kwasu foliowego, tiaminy, witaminy C, koenzymu Q10, kortykosteroidów itp.) Również nie zahamowało postępu tej patologii..
Najczęściej stosuje się metody medyczne skoncentrowane na kontroli objawów i opiece paliatywnej.
Niezbędne jest zarządzanie objawami podmiotowymi i przedmiotowymi przez multidyscyplinarny zespół medyczny: okuliści, nefrolodzy, endokrynolodzy, neurolodzy, kardiolodzy itp..
Zespół MELAS ma zwykle przebieg określany jako nawrót, remisja lub prezentacja ostrych ataków, co utrudnia dokładną ocenę skuteczności nowych podejść terapeutycznych.
Pacjenci dotknięci chorobą nieuchronnie rozwijają upośledzenie funkcji poznawczych, zaburzenia psychomotoryczne, utratę wzroku i słuchu oraz inne komplikacje medyczne aż do śmierci.



Jeszcze bez komentarzy