Plik Zespół Pierre Robina Jest to schorzenie pochodzenia genetycznego zaliczane do zespołów lub patologii twarzoczaszki. Klinicznie charakteryzuje się mikrognatią, glossoptozą, niedrożnością górnych dróg oddechowych i zmienną obecnością rozszczepu podniebienia..
Jeśli chodzi o etiologiczne pochodzenie tej patologii, zespół Pierre-Robena wynika z obecności określonych mutacji w genie SOX9, przy czym większość przypadków jest zdiagnozowana.
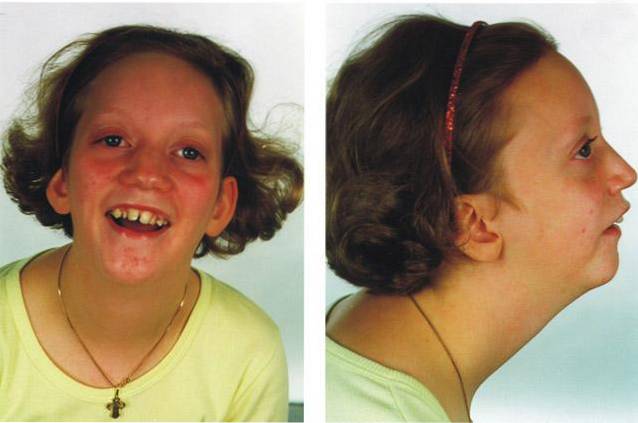
Ogólnie rzecz biorąc, zespół ten powoduje poważne komplikacje medyczne, w tym niewydolność oddechową, zwierzęta trawienne lub rozwój innych wad rozwojowych twarzoczaszki..
Z drugiej strony rozpoznanie zespołu Pierre-Robina zwykle nie zostaje potwierdzone do momentu urodzenia; Oprócz wyników klinicznych konieczne jest przeprowadzenie różnych badań radiologicznych w celu zidentyfikowania zmian kostnych..
Obecnie nie ma lekarstwa na zespół Pierre Robin, jednak w celu skorygowania nieprawidłowości układu mięśniowo-szkieletowego często stosuje się podejścia chirurgiczne. Ponadto, aby uniknąć powikłań medycznych zagrażających życiu, ważne są adaptacje układu oddechowego i żołądkowo-jelitowego..
Indeks artykułów
Zespół Pierre Robin to wrodzona patologia, której objawy kliniczne są obecne od momentu urodzenia, a ponadto wszystkie jego cechy są związane z obecnością wady rozwojowej twarzoczaszki.
Ponadto w literaturze medycznej możemy zidentyfikować różne terminy używane w kontekście zespołu Pierre Robin: choroba Pierre Robin, malformacja Pierre Robin czy sekwencja Pierre Robin.
Na określonym poziomie zespół ten został po raz pierwszy opisany w 1891 roku przez Menerad i Lannelongue. W doniesieniach klinicznych opisali dwóch pacjentów, których przebieg kliniczny charakteryzował się niedorozwojem struktury kości żuchwy, rozszczepem podniebienia oraz przemieszczeniem lub retrakcją języka..
Jednak dopiero w 1923 roku Pierre Robin w pełni opisał kliniczne spektrum tej patologii, skupiając się w swoich badaniach na przypadku dziecka dotkniętego wadą rozwojową żuchwy, nieprawidłowo dużym językiem i poważnymi problemami z oddychaniem..
Pomimo tego, że patologię tę zasadniczo wyróżnia badanie radiologiczne twarzoczaszki, charakteryzuje się dużą ruchliwością związaną z powikłaniami medycznymi, głównie związanymi z niewydolnością serca i problemami z karmieniem..
W szczególności zespół Pierre Robin ma wysoką śmiertelność związaną z niedrożnością dróg oddechowych, nieprawidłowościami neurologicznymi lub zaburzeniami pracy serca.
Z drugiej strony wielu autorów woli nazywać tę patologię jedynie sekwencją Pierre'a, ponieważ to anomalie żuchwy mają tendencję do wywoływania pozostałych typowych oznak i objawów..
Częstość występowania zespołu Pierre Robin szacuje się na około jeden przypadek na każde 8500 dzieci urodzonych żywcem, z czego ponad 80% zdiagnozowanych przypadków wiąże się z innymi powikłaniami medycznymi i określonymi zespołami.
Z drugiej strony, w przypadku Stanów Zjednoczonych zapadalność na zespół Pierre Robin wynosi 1 przypadek na 3120 urodzeń każdego roku.
Obecnie nie zidentyfikowano również zróżnicowanej częstości występowania zespołu Pierre Robin związanego z płcią, geografią lub określonymi grupami etnicznymi i rasowymi..
Ponadto, jak wskazaliśmy wcześniej, zespół Pierre Robin jest jedną z patologii twarzoczaszki o wysokim prawdopodobieństwie zgonu. W Stanach Zjednoczonych około 16,6% osób dotkniętych chorobą umiera z powodu rozwoju powikłań medycznych.
W kolejności występowania najczęstszymi wtórnymi patologiami medycznymi są: wady serca (39%), zmiany w ośrodkowym układzie nerwowym (33%) i anomalie w innych narządach (24%).
Sekwencję Pierre Robina różni się od innych typów patologii skóry twarzoczaszki obecnością trzech podstawowych cech klinicznych: mikrognatii, glossoptozy i rozszczepu podniebienia:
Terminem mikrognathia mówimy o obecności patologicznej zmiany w rozwoju struktury żuchwy, a konkretnie ostateczny kształt ma mniejszy rozmiar w porównaniu z oczekiwanym dla poziomu rozwoju osoby dotkniętej chorobą..
W konsekwencji niepełny rozwój tej struktury twarzoczaszki spowoduje wiele różnorodnych zmian, z których wszystkie związane są z obecnością wad rozwojowych ust i twarzy..
Mikrognathia jest objawem medycznym występującym u około 91% osób dotkniętych zespołem Pierre Robin..
Przez termin glossoptosis odnosimy się do obecności nieprawidłowego cofnięcia się języka w obrębie struktury jamy ustnej, w szczególności, języki mają tendencję do umieszczania dalej niż normalnie w wyniku mikrografii i zmniejszenia objętości jamy ustnej. Jama ustna.
Nieprawidłowości związane z pozycją i strukturą języka mogą powodować poważne problemy z karmieniem, które mogą prowadzić do poważnych schorzeń..
Ponadto w innych przypadkach można również zidentyfikować nienormalnie duży język (makroglossia), który utrudnia między innymi oddychanie, żucie lub wytwarzanie funkcjonalnego języka..
Ponadto glosoptoza jest jednym z najczęstszych objawów klinicznych zespołu Pierre Robina, obserwowanym w około 70-85% zdiagnozowanych przypadków. Podczas gdy makroglossię można zaobserwować w mniejszym odsetku, u około 10-15% osób dotkniętych chorobą.
Termin ten odnosi się do obecności wady rozwojowej w okolicach podniebienia lub dachu policzkowego, czyli można zaobserwować obecność pęknięć lub dziur związanych z niepełnym rozwojem żuchwy..
Podobnie jak w przypadku innych wyników klinicznych, rozszczep podniebienia spowoduje istotne zmiany w żywieniu.
Oprócz tych oznak i objawów można również zidentyfikować inne rodzaje zmian, w tym:
- Wady rozwojowe nosa.
- Zaburzenia oka.
- Zmiany i wady rozwojowe układu mięśniowo-szkieletowego, głównie związane z rozwojem oligodaktyli (zmniejszenie liczby palców, mniej niż 5 w dłoniach lub stopach), klinodaktylia (poprzeczne odchylenie ułożenia palców), polidaktylia (zwiększona liczba palców), hipermobilność w stawach (nienormalnie przesadny wzrost ruchomości stawów), dysplazja w paliczkach (paliczki ze słabym lub niepełnym rozwojem kości) lub syndaktylia (zespolenie kilku palców).
- Inne zmiany: można również zidentyfikować wady rozwojowe w budowie kończyn lub kręgosłupa.
Oprócz funkcji medycznych opisanych powyżej mogą pojawić się inne związane z różnymi systemami:
Zmiany kardiologiczne stanowią jedno z powikłań medycznych o największym wpływie na zdrowie jednostki, stwarzające istotne zagrożenie dla jego przeżycia. Jednak oznaki i objawy związane z układem sercowo-naczyniowym można zwykle leczyć metodami farmakologicznymi i / lub chirurgicznymi..
Niektóre z najczęstszych nieprawidłowości w sercu obejmują zwężenie mięśnia sercowego, uporczywy otwór owalny, zmienione tętnice przegrody lub nadciśnienie..
Genetyczne pochodzenie zespołu Pierre Robin może również oznaczać rozwój różnych zmian neurologicznych, związanych głównie z obecnością nieprawidłowości w ośrodkowym układzie nerwowym (OUN)..
Dlatego niektóre z zaburzeń neurologicznych najbardziej związanych z zespołem Pierre Robin mogą obejmować wodogłowie, malformację Chiariego, epizody epilepsji lub opóźnienia w nabywaniu umiejętności psychomotorycznych..
Zmiany w oddychaniu są jedną z najważniejszych cech, ponieważ mogą powodować zarówno śmierć pacjenta z powodu niewydolności oddechowej, jak i rozwój uszkodzenia mózgu z powodu braku tlenu w obszarach nerwowych.
Dlatego też w wielu przypadkach konieczne są korekty chirurgiczne w celu udrożnienia dróg oddechowych, głównie korekcja dysplazji żuchwy czy ustawienia języka..
Podobnie jak w przypadku chorób układu oddechowego, problemy z karmieniem wynikają zasadniczo z wad rozwojowych żuchwy.
Dlatego już od urodzenia istotne jest rozpoznanie tych nieprawidłowości, które utrudniają karmienie, aby je skorygować, a tym samym zmniejszyć prawdopodobieństwo wystąpienia chorób związanych z niedożywieniem..
Zespół lub sekwencja Pierre Robina ma podłoże genetyczne i jest związane ze zmianami w genie SOX9. Chociaż tę anomalię zidentyfikowano w większości izolowanych przypadków zespołu Pierre Robin, niektóre jej cechy kliniczne mogą być związane z innymi typami mutacji pochodzenia genetycznego..
W szczególności gen SOX9 odgrywa podstawową rolę w dostarczaniu niezbędnych instrukcji biochemicznych do produkcji białka zaangażowanego w rozwój i tworzenie różnych tkanek i narządów podczas rozwoju płodowego..
Ponadto obecne badania wskazują, że białko SOX9 może regulować aktywność innych typów genów, zwłaszcza zaangażowanych w rozwój struktury szkieletowej, a tym samym żuchwy..
W efekcie zmiany genetyczne uniemożliwiają prawidłowy rozwój morfologiczny pewnych struktur, a zatem pojawiają się kardynalne objawy kliniczne: mykognatia, glossoptoza i rozszczep podniebienia..
W wielu przypadkach wady strukturalne twarzoczaszki można wykryć w czasie ciąży za pomocą USG, chociaż przypadki są rzadkie.
W tym sensie podejrzenie zespołu Pierre Robin jest bardziej powszechne w okresie poporodowym lub niemowlęcym. U większości chorych objawy strukturalne są wyraźnie widoczne, dlatego rozpoznanie jest potwierdzane badaniami radiologicznymi połączonymi z badaniem przedmiotowym..
Jednak w drugim przypadku konieczne jest wcześniejsze przeprowadzenie badania układu oddechowego, a następnie badania radiologicznego w celu ustalenia obecności tego zespołu..
Ponadto innym fundamentalnym aspektem w diagnostyce tej patologii jest eksploracja innych obszarów, zwłaszcza układu sercowo-nerwowego, ponieważ mogą pojawić się inne rodzaje anomalii zagrażających życiu..
Wreszcie, interwencja diagnostyczna może obejmować indywidualne i rodzinne badanie genetyczne w celu zidentyfikowania możliwych powiązań genetycznych..
Typowe leczenie zespołu Pierre Robin opiera się na zabiegach chirurgicznych korygujących wady rozwojowe twarzoczaszki:
- Tracheotomia.
- Zamknięcie szczeliny podniebiennej.
- Wydłużenie szczęki.
- Rozkojarzenie kości.
- Fiksacja językowa.
Ponadto w leczeniu patologii serca, epilepsji i innych zdarzeń neurologicznych stosuje się również inne podejścia farmakologiczne..
Ponadto osoby dotknięte chorobą mają zwykle trudności związane z produkcją języka, dlatego w wielu przypadkach wczesna terapia logopedyczna jest niezbędna..
Podstawowym celem jest ustanowienie skutecznej metody komunikacji poprzez pozostałe zdolności, a co za tym idzie, stymulowanie nabywania nowych umiejętności..



Jeszcze bez komentarzy